
The Townsend Lab is located in the Cancer-Cardiovascular Research Building.
Research Projects
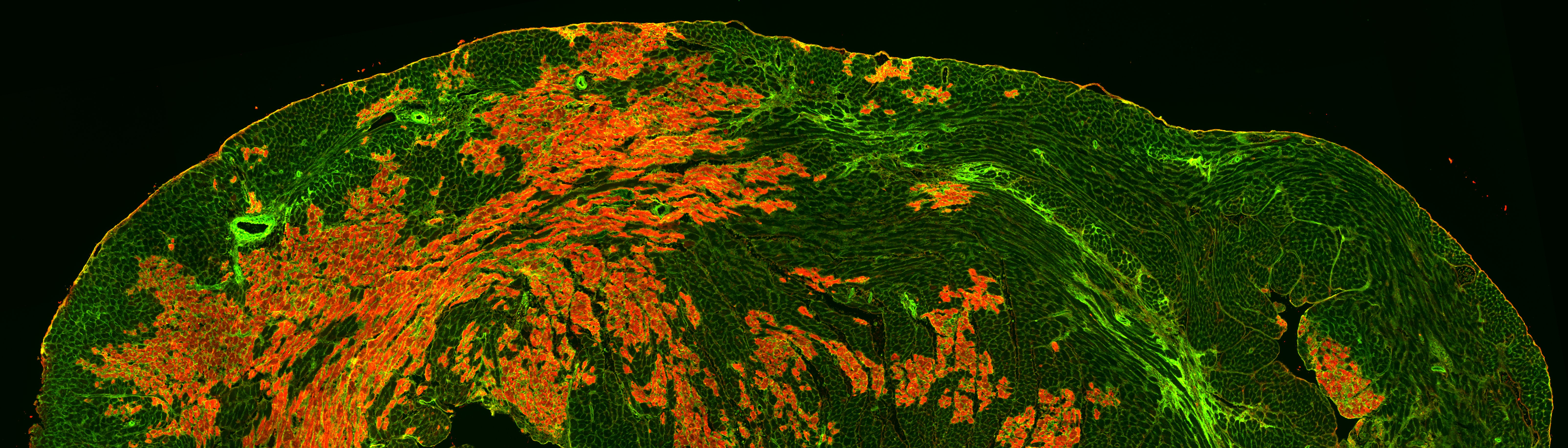
Preventing Myocyte Loss
Dystrophin Replacement
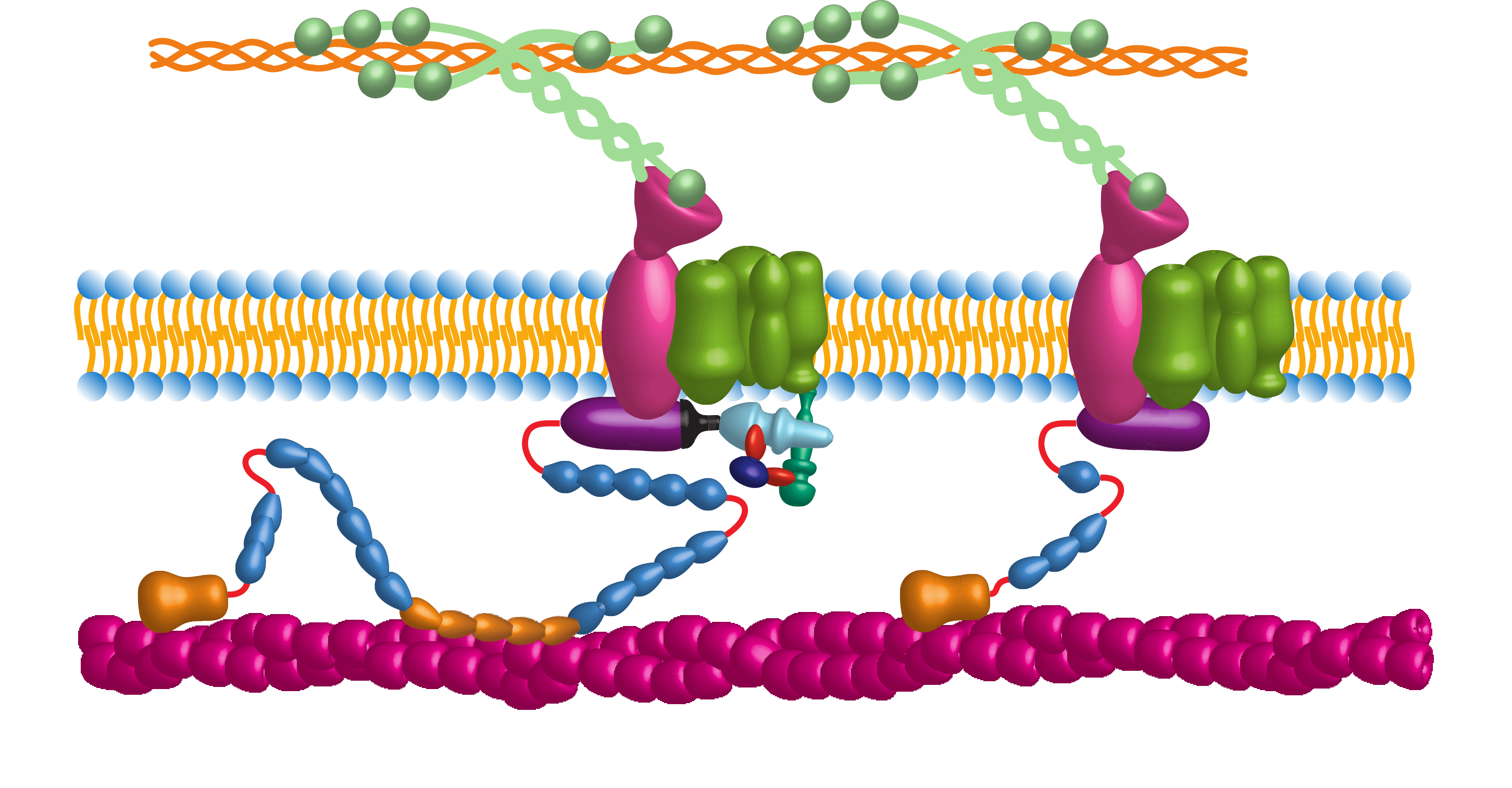
Dystrophin is a very large protein which makes it difficult to replace. However, it turns out that its possible to avoid the worst of DMD with only a small fraction of the whole protein. The goal of this project is to understand which domains are critical for protecting the heart.
Blocking Angiotensin II Signaling
Angiotensin II is a hormone that normally helps regulate your blood pressure, but it also plays a role in damaging dystrophic hearts. It turns out that blocking the angiotensin II receptor protects the dystrophic heart from injury during stress. Where this angiotensin II comes from and how it works may provide new therapies for slowing the progression of dystrophic cardiomyopathy.
Preventing Membrane Damage
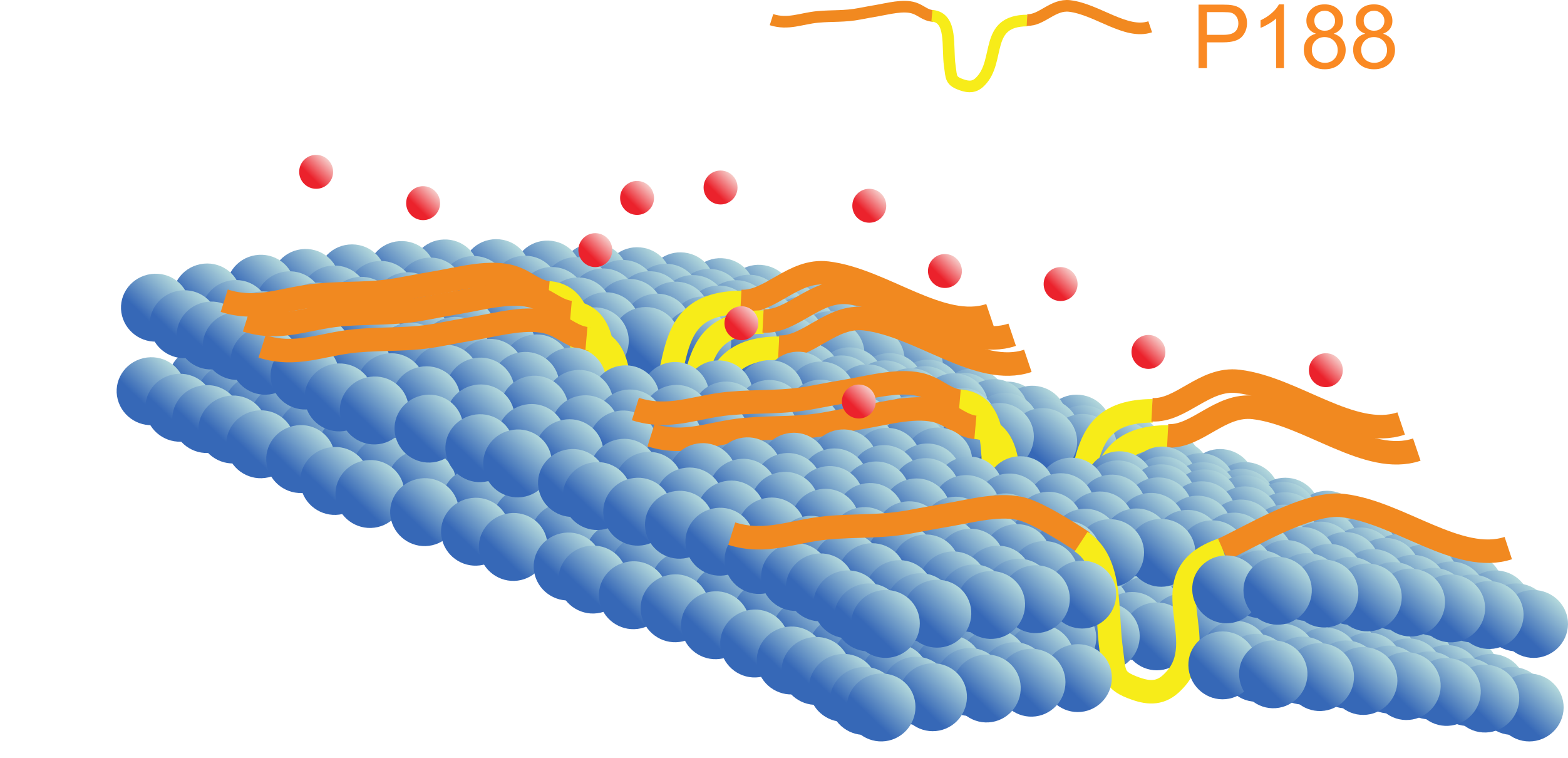
Many forms of dystrophic cardiomyopathy are characterized by leaky membranes. One potential therapy is to reinforce the membrane with a chemical sealant. These "molecular bandaids" help protect the dystrophic heart from injury.
Promoting Cell Survival
Maintain Cellular Energetics
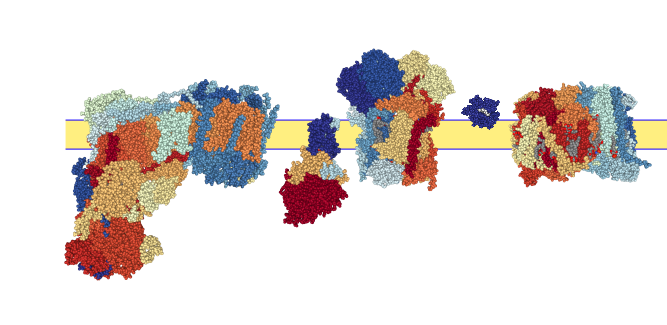
The dystrophic heart has less energy than a healthy heart. We think this limits the heart's response to stress and contributes to myocyte death. By understanding the causes for this reduced energy production, we hope to help these cells survive even if they are injured.
Protect the Mitochondria
When the heart works harder more Ca2+ is released with each beat. This extra Ca2+ enters the mitochondria and super charges ATP production. However, if Ca2+ gets too high it can cause mitochondria to shutdown, stopping energy production and maybe taking the whole cell down with it. In the dystrophic heart, Ca2+ is already high at rest. When the heart beats harder, the increased Ca2+ may be too much for the mitochondria in the heart cell. We are interested in exploring how this might affect the dystrophic heart.
Prevent Energetic Catastrophe
Millions of years ago our ancestral cells made a deal with a plucky little microbe. Through the years of evolutionary negotiations our cells and this microbe have come to an occasionally uneasy truce. In return for protection and a steady source of oxygen and reduced carbon these mitochondria provide our cells with ATP (and other metabolic products). While our cells can end a mitochondria through mitophagy, mitochondria can initiate the death of our cells. In the dystrophic heart this relationship is stressed. Often mitochondria are provoked to contribute to the irreversible loss of a cardiomyocyte. These studies look to counteract the destabilizing effects of dystrophy on cellular-mitochondrial relationship to maintain a balance that preserves myocardial function.
Limiting the Damage
Calming the Immune System
Disruption of the membrane results in the release of intracellular contents into the extracellular space. Like sharks sensing blood in the water, immune cells are drawn to this cytosolic bleeding. It is possible that these cells, in their haste to clean up cellular debris, may accelerate the death of other myocytes in the area. Understanding how these immune cells contribute to myocardial injury may provide new targets to preserve dystrophic myocardium.
ROS and the Dystrophic Heart
The addition of a single electron to molecular oxygen creates a superoxide (O2-). Under normal circumstances this is rapidly converted to hydrogen peroxide (H2O2) by superoxide dismutase and this H2O2 is converted to water and oxygen by catalase. If these defenses are overwhelmed than these reactive oxygen species (ROS) participate in other reactions that create molecules capable of ripping electrons away from carbons in the lipid bilayer. There are several lines of evidence indicating that ROS contributes to the pathology in the dystrophic heart. These studies aim to better understand how this may happen and how to prevent it.